清华大学物理系徐勇、段文晖研究组将深度学习电子结构方法拓展至杂化密度泛函计算
近日,清华大学物理系徐勇、段文晖研究组与合作者发展了深度学习计算方法,将研究组开发的深度学习密度泛函哈密顿量(DeepH)方法从半局域密度泛函推广至杂化密度泛函。这一进展使DeepH能够兼容更高精度的第一性原理计算方法。鉴于杂化密度泛函对激发态性质描述更为准确,此方法提升了深度学习电子结构计算的精度,并有望实现深度学习驱动的高精度光学性质计算。
伴随着人工智能领域的发展,深度学习与第一性原理计算的结合展示出加速材料计算模拟、推动人工智能驱动的材料发现的巨大潜力,已成为计算物理和计算材料学研究的前沿热点之一。DeepH方法是深度学习电子结构计算的代表性方法,其旨在跳过密度泛函理论(DFT)的自洽求解过程,直接预测DFT的核心物理量——DFT哈密顿量。在维持亚-毫电子伏特级别的高精度前提下,DeepH方法实现了高效率、低标度、可泛化的DFT哈密顿量预测。这一方法被陆续拓展至磁性体系、电声耦合等物理问题中,并展现出构建“材料大模型”的潜力。然而,已有的DeepH研究主要集中于广义梯度近似等半局域DFT近似泛函,这类近似存在系统性低估半导体带隙等问题。杂化密度泛函通过引入Hartree-Fock精确交换相互作用,对半导体带隙等激发态性质的描述更为准确,但计算代价较高。将DeepH方法推广至杂化密度泛函,有望突破杂化密度泛函的计算效率瓶颈,将这种高精度第一性原理计算方法应用于大体系、高通量材料计算模拟中。
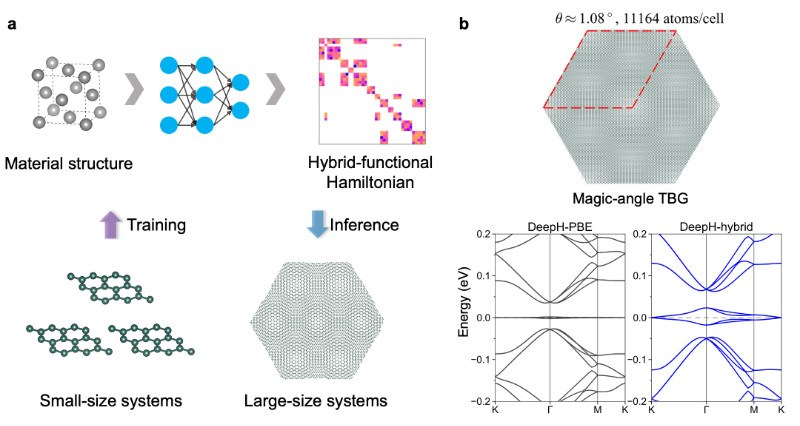
在最新的研究中,研究团队开发了适用于杂化密度泛函的DeepH方法DeepH-hybrid(图a)。DeepH方法的基础是“量子近视性”原理,即材料局部的哈密顿量只与一定半径内的材料结构相关。研究团队指出,尽管杂化密度泛函包含非局域的精确交换相互作用,但同样具备间接的“量子近视性”,因此DeepH框架仍适用于处理杂化密度泛函哈密顿量的深度学习;针对这种修正的“量子近视性”,研究团队调整了网络的建图半径,并在石墨烯等多个测试体系上均达到了亚-毫电子伏特精度。DeepH-hybrid方法跳过了杂化密度泛函中耗时的自洽迭代、哈密顿量对角化、精确交换相互作用求解等过程,直接预测杂化密度泛函哈密顿量,计算效率相比传统杂化密度泛函提升多个数量级,且关于材料的尺度有线性的时间复杂度。基于DeepH-hybrid方法的高效性,研究团队开展了多个实例研究,包括莫尔转角材料魔角石墨烯(图b),其超胞内有超过1万个原子。研究结果表明,在魔角石墨烯的关联平带的能带结构方面,杂化密度泛函与半局域密度泛函的结果有定性不同,如平带的带宽存在数量级的差异,预示着在莫尔转角体系中,精确交换相互作用的引入可能对计算结果有重要影响。本工作的杂化密度泛函数据由国产第一性原理计算软件ABACUS产生,DeepH已经支持对接ABACUS程序。
目前,深度学习与第一性原理计算的交叉领域正在迅速发展,基于材料大数据和人工智能的材料发现变得可行。基于材料大数据,有望开发预测任意材料性质的DeepH通用材料模型。在这一背景下,基于杂化密度泛函等更高精度第一性原理计算的材料大数据,将为开发对应的更精确DeepH通用材料模型提供坚实基础,进而在本工作的基础上,推动更精准的深度学习材料发现。
相关研究成果以“一种有效的混合密度泛函计算的深度等变神经网络方法”(A deep equivariant neural network approach for efficient hybrid density functional calculations)为题,于10月11日发表于《自然·通讯》(Nature Communications)。
清华大学物理系教授徐勇、段文晖,中国科学院物理研究所特聘研究员任新国为论文的共同通讯作者,清华大学物理系2023级博士生唐泽宸、已毕业博士李贺,合肥综合性国家科学中心人工智能研究院副研究员林霈泽为论文共同第一作者。合作者还包括北京大学化学与分子工程学院研究员蒋鸿、中国科学技术大学物理学院教授何力新、清华大学物理系访问学生贡晓荀以及合肥综合性国家科学中心人工智能研究院博士后金敢。研究得到国家自然科学基金委基础科学研究中心、国家科技部重点研发计划、国家自然科学基金重点项目、北京市未来芯片技术高精尖创新中心、北京材料基因工程高精尖创新中心、天津超算中心等项目单位的支持。