新改进的量子算法能执行full-CI计算,无需控制时间演化
为了不断改进之前的工作,大阪城市大学研究生院的一个研究小组应用其最近开发的贝叶斯相位差估计(BPDE)量子算法来执行原子和分子的全组态相互作用(full-CI)计算,它不需要模拟波函数随辅助量子比特的时间演化。在量子计算中,与传统的量子门并行执行方法相比,这种新算法更易于在实际的量子计算机中实现。
大阪市立大学理科研究生院的研究人员继续改进了他们最近开发的量子算法,这一次将其应用于计算氢分子在无需控制时间演化下的势能曲线。通过在他们之前开发的BPDE量子算法中加入受控态准备的步骤,该团队绕过了模拟波函数随时间演化时需要的辅助量子比特条件。他们的研究作为开放获取文章发表在近期的《物理化学快报》上。
这解决了传统量子算法里的一个常见问题,即量子门和许多非相邻的双量子比特量子门的并行处理,并证明其自身是一种可在量子计算机上执行的量子算法,该算法用来执行原子和分子的全组态相互作用计算。全组态相互作用(full-CI)计算能够为微观系统提供薛定谔方程的优化解,但由于所需的计算时间呈指数级增长,因此对于经典计算机而言是难以处理的。
在精确求解薛定谔方程以了解原子或分子的电子态的竞赛中,科学家们已转向量子计算机,以在多项式时间内进行化学计算。量子相位估计(QPE)算法已成为众所周知的强大工具,可用于对小分子的波函数进行全组态相互作用计算,并且科学家们已经进行了各种尝试。使基于QPE的方法能够解决以指数级累积的计算成本,它与所研究的系统规模相对应。
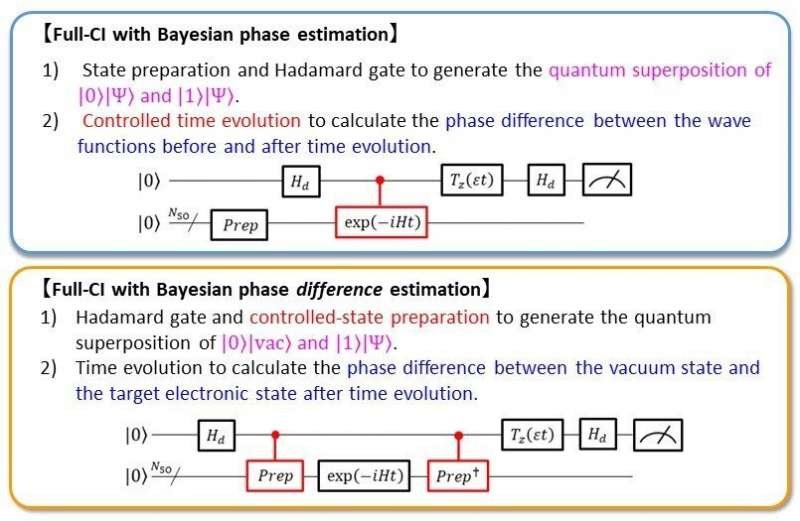
该研究主要作者、大阪城市大学特聘讲师Kenji Sugisaki说:“基于QPE的方法来模拟辅助量子比特上波函数的时间演化,这需要许多受控量子门,其存在也阻碍了这些门的并行执行和量子电路的压缩。在我们的这项研究中,应用了我们的贝叶斯相位差估计(BPDE)算法,它是QPE的一种改进版,能够绕过受控时间演化操作的需要来执行全组态相互作用计算。”
研究顾问Kazunobu Sato教授说:“仅当第一个量子比特处于|1⟩态时才演化波函数,这使得量子门难以并行化。我们重写了量子逻辑电路,以便无论第一个量子比特处于|0⟩还是|1⟩态都可以应用时间演化算子,这将允许轻松并行处理量子门,这增加在实际量子计算机中实现该算法的可能性。”
为此,该团队引入了一种受控态准备,它构建了具有零电子的“真空”波函数|vac⟩和目标电子态的波函数|Ψ⟩的量子叠加。其逻辑电路是(|0⟩|vac⟩+|1⟩|Ψ⟩)⁄√2。该大学名誉教授Takeji Takui解释说:“换句话说,我们计算了一个原子或分子的全组态相互作用能量作为其电离能。”
该团队通过计算氢分子的四个价电子态的势能曲线,举例说明了他们基于BPDE的全组态相互作用计算方法的效率。重要的是,不受受控时间演化的限制使得量子门的并行化和在真实量子设备上实现它要更容易,这让团队希望他们的贝叶斯相位差估计算法为更实用的full-CI计算铺平道路,并成为精确量子化学的代名词。(编译:Qtech)