日本研究人员开发BPDE量子算法,通过计算能量差了解电子状态
大阪市立大学科学研究生院的研究人员开发了一种新量子算法,可以通过直接计算原子或分子系统相关态的能量差来了解它们的电子状态。该算法打破常规,应用的贝叶斯相位差的不同估计,它不关注从相位前和相位后演化计算的总能量差异,而是跟踪能量差异本身的演化。
该研究负责人兼大阪市立大学特聘讲师Kenji Sugisaki说:“几乎所有的化学问题都讨论能量差异,而不是分子本身的总能量。此外,出现在元素周期表下部的重原子分子具有较大的总能量,但化学中讨论的能量差异的大小(例如电子激发态和电离能)与分子的大小无关。”该想法促使Sugisaki和他的团队实施了一种直接计算能量差异而不是总能量的量子算法,这创造了一个利用可扩展或实用的量子计算机使我们能够进行实际化学研究和材料开发的未来。
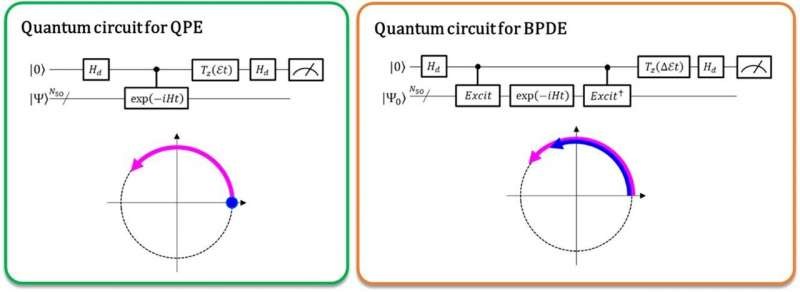
目前,量子计算机能够执行全组态相互作用(full-CI)计算,通过被称为量子相位估计(QPE)的量子算法提供最佳分子能量,并且任何超级计算机对于相当大的分子系统是难以进行full-CI计算的。QPE依赖于这样一个事实:波函数|Ψ⟩,这是用来表示微观系统量子态的数学描述,在这种情况下,微观系统(如原子或分子)的薛定谔方程的数学解会随着时间眼花,并根据它的总能量改变它的相位。在传统的QPE中,量子叠加态(|0⟩|Ψ⟩+|1⟩|Ψ⟩)⁄√2,并且引入了受控时间演化算子使得|Ψ⟩仅在第一个量子位为|1⟩态时才随时间演化。因此,|1⟩态在时间上创造了进化后的量子阶段,而|0⟩态创造了进化前的量子阶段。进化前和进化后之间的相位差给出了系统的总能量。
大阪市立大学的研究人员将传统的QPE推广到直接计算两个相关量子态之间总能量的差异。在新实现的被称为“贝叶斯相位差估计”(BPDE)的量子算法中,两个波函数的叠加(|0⟩|Ψ₀⟩+|1⟩|Ψ₁⟩)⁄√2,其中|Ψ ₀⟩和|Ψ₁⟩分别表示与各态相关的波函数已准备好,|Ψ₀⟩和|Ψ₁⟩之间的相位差叠加后的时间演化直接给出了所涉及的两个波函数之间总能量差异。该研究主管兼名誉教授Takeji Takui说:“我们强调,该算法遵循能量差随时间的演变,这比单独计算原子或分子的总能量更不容易产生噪音。因此,该算法适合需要精确能量高精确度的化学问题。”
此前,该研究小组开发了一种量子算法,可以直接计算具有不同自旋量子数的电子态(自旋态)间的能量差异。然而,那种算法比传统的QPE需要更多的量子比特,并且不能应用于具有相同自旋量子数的电子态间的能量差计算,这对于紫外/可见吸收光谱(都属于分子光谱)的光谱分配很重要。新研究中开发出的BPDE算法克服了这些问题,使其成为了一种高度通用的量子算法。(编译:Qtech)